
Table of Contents
Benefits and Challenges of eSTAR
Leveraging Industry Expert Support

eSTAR Overview
What Is The FDA’s eSTAR Program?
The FDA eSTAR (Electronic Submission Template and Resource) Program is a tool designed to improve the efficiency and consistency of medical device submissions. It is a user-friendly, interactive PDF template that guides applicants through the process of submitting premarket notifications, known as 510(k)s, to the FDA. De Novo requests, pre-submissions (formally known as q-submissions), and PMAs may also be submitted to the FDA using eSTAR. By digitizing the submission process, eSTAR ensures that applicants provide all required information, improving the quality of submissions and reducing review time.
The eSTAR template aligns with regulatory requirements and includes automated cross-checks and prompts, helping manufacturers avoid common errors. It covers all major sections needed for FDA review, from device description and labeling to risk analysis and clinical data.
The FDA eSTAR program is part of the agency’s ongoing efforts to digitize and streamline its review processes, improving transparency and communication between the FDA and device manufacturers.
History of eSTAR
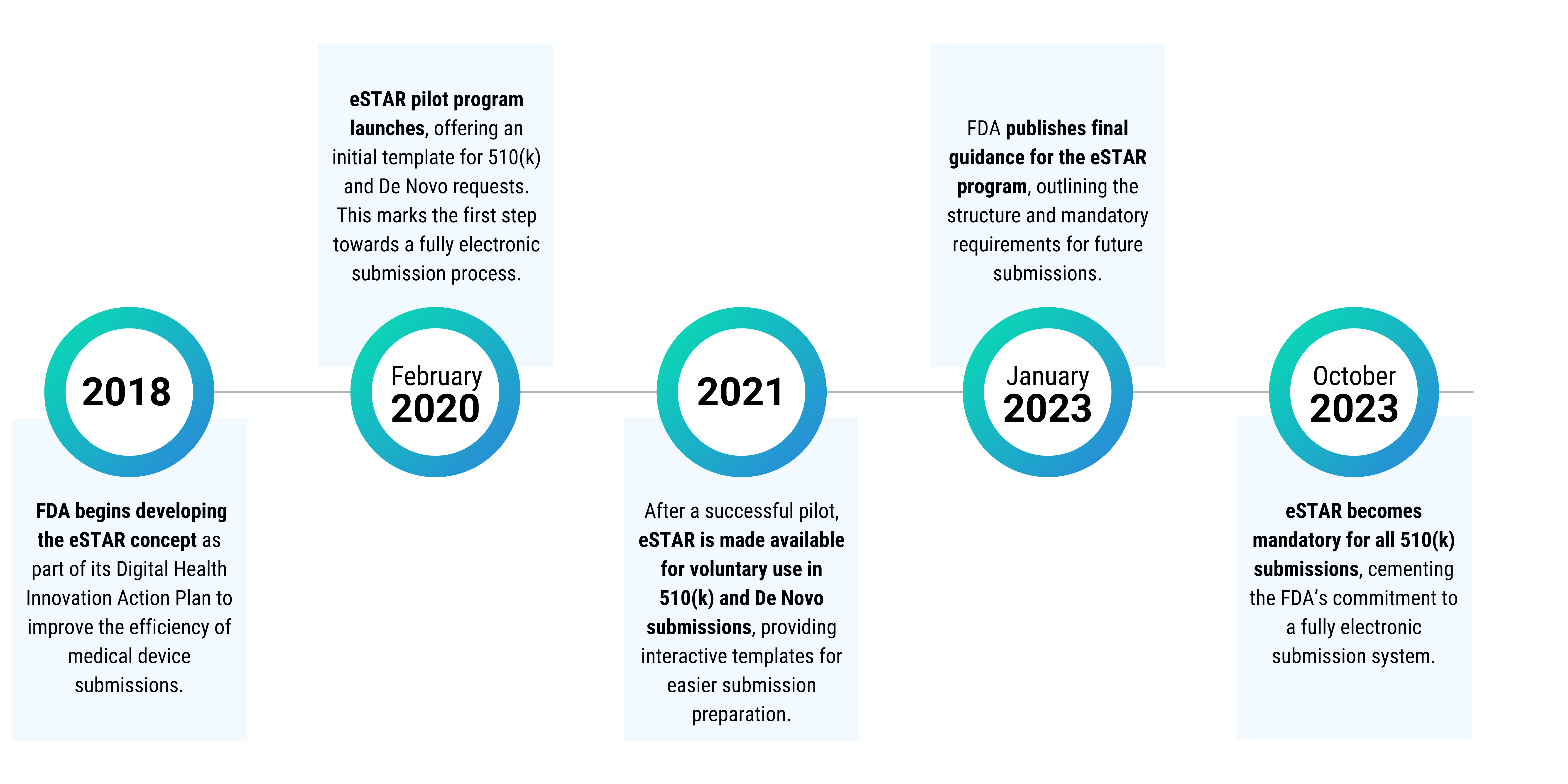
The eSTAR program was launched as part of the FDA’s broader commitment to modernize regulatory processes and adapt to the increasing complexity of medical device submissions. It began as a pilot program in February 2020, aimed at addressing inconsistencies in submissions, which often caused delays due to missing or incomplete information.
In October 2021, after a successful pilot, the FDA officially made eSTAR available for voluntary submissions for 510(k) and De Novo requests. This marked a shift towards more streamlined and consistent submission practices. The eSTAR program built on the foundation laid by earlier digital initiatives, such as the eCopy program, which required electronic versions of submissions but lacked the interactivity and real-time feedback offered by eSTAR.
The development of eSTAR aligns with the FDA’s broader Digital Health Innovation Action Plan and Medical Device User Fee Amendments (MDUFA), which emphasize leveraging technology to improve the speed and predictability of the regulatory process.
Purpose of eSTAR
The eSTAR program aims to improve the quality of submissions for various medical devices by guiding submitters to provide thorough and well-organized data for premarket review.
The main goal of eSTAR is to simplify and standardize the submission process for medical devices, ultimately shortening review times and decreasing the likelihood of submission rejections. The program addresses common issues in the traditional submission process, such as incomplete or inconsistent documentation, and improves the overall quality of submissions.
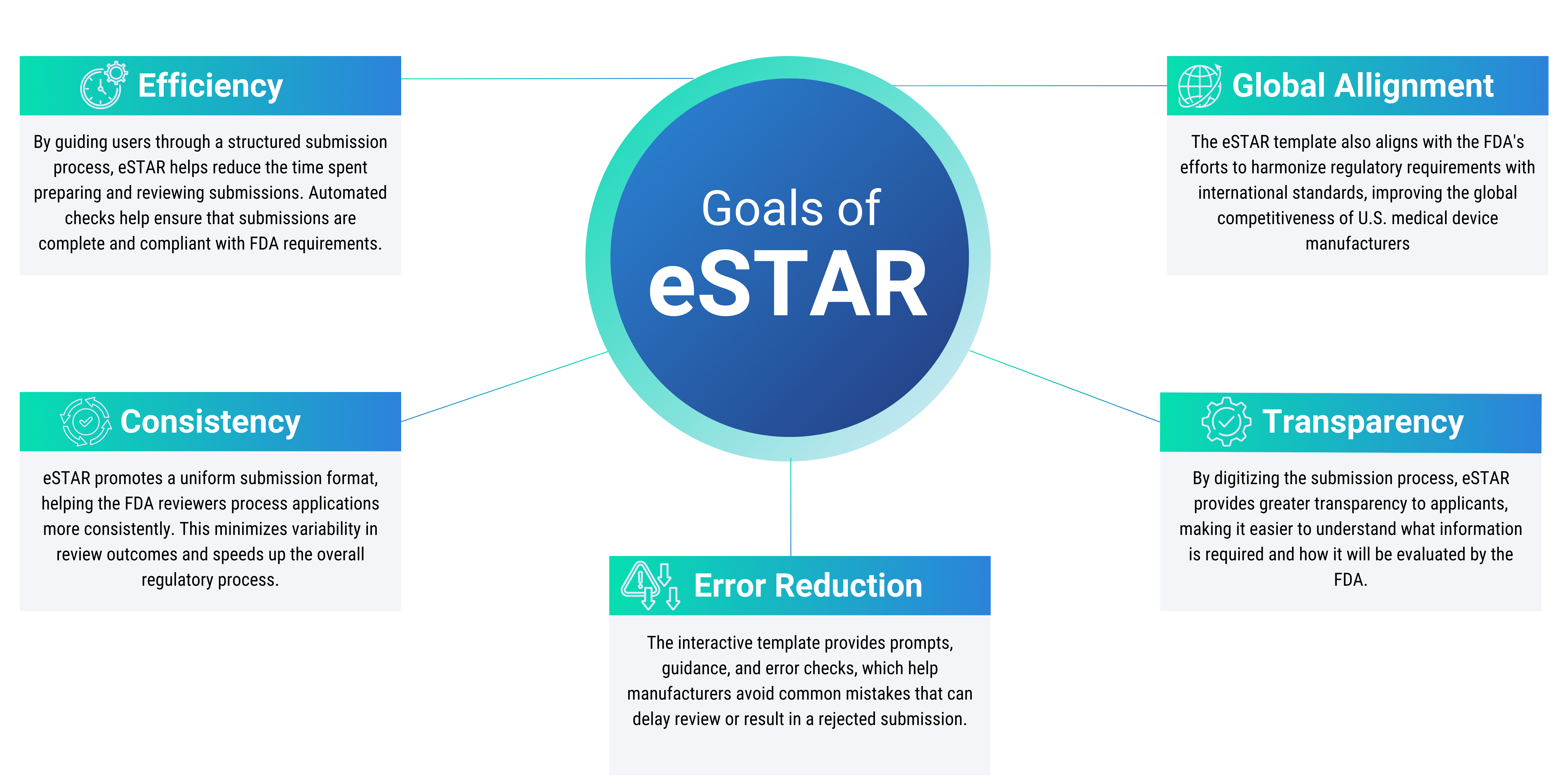
Goals of eSTAR
The eSTAR program aims to streamline the FDA submission process by enhancing efficiency, consistency, and transparency. It guides users through a structured process with automated checks, reducing preparation time and minimizing errors. eSTAR promotes a uniform submission format for consistency in reviews and aligns with international regulatory standards, ultimately making the approval of medical devices more predictable and accessible.
eSTAR Users and Fees
Who Needs to Submit Through eSTAR?
As of October 2023, the FDA has made it mandatory for all medical device manufacturers submitting 510(k) premarket notifications to use eSTAR, unless exempted. This requirement applies to both traditional and abbreviated 510(k) submissions. Manufacturers seeking to bring a new device to market or modify an existing device must use eSTAR to streamline their submission and ensure compliance with the FDA’s regulatory standards.
In addition to 510(k) submissions, eSTAR can also be used for voluntary De Novo requests, which are required for devices that are novel and not substantially equivalent to any existing devices on the market. It should be noted that the final guidance Electronic Submission Template for Medical Device De Novo Requests requires that all De Novo submissions must use eSTAR unless exempted starting October 1, 2025.

Manufacturers of various types of medical devices—from diagnostic tools to complex therapeutic technologies—are encouraged to adopt eSTAR to ensure their submissions are complete, reduce errors, and expedite the review process. This system is designed to facilitate a smoother regulatory pathway for both U.S.-based and international manufacturers seeking FDA approval for their products.
Exemptions
As described in the FDA guidance, “Electronic Submission Template for Medical Device 510(k) Submissions,” the following 510(k) submissions/information are exempted from the electronic submission requirements:
- Interactive Review Responses
- Amendments:
- Appeals/Requests for Supervisory Review
- Substantive Summary Requests
- Change in Correspondent Amendments
- Amendments After Final Decision (i.e., add-to-files)
- Withdrawal Requests
User Fees
My Section
When submitting through the FDA’s eSTAR program, the applicable user fee must be paid for the overall submission, not just for using eSTAR. The fee corresponds to the type of premarket submission and must be processed before submission. For 510(k) submissions, whether traditional or abbreviated, manufacturers are required to pay the FDA’s 510(k) user fee, which is based on the fiscal year and company size (standard or small business).
For voluntary De Novo requests submitted via eSTAR, manufacturers are required to pay the De Novo user fee. Ensuring correct fee payment is crucial to avoid delays in the FDA review process. Payment of these fees is required at the time of submission, and the FDA’s website provides up-to-date fee schedules and small business discount eligibility information. n.
Below is a table detailing the FDA’s user fees for each submission type for FY 2025, effective October 1st, 2024.
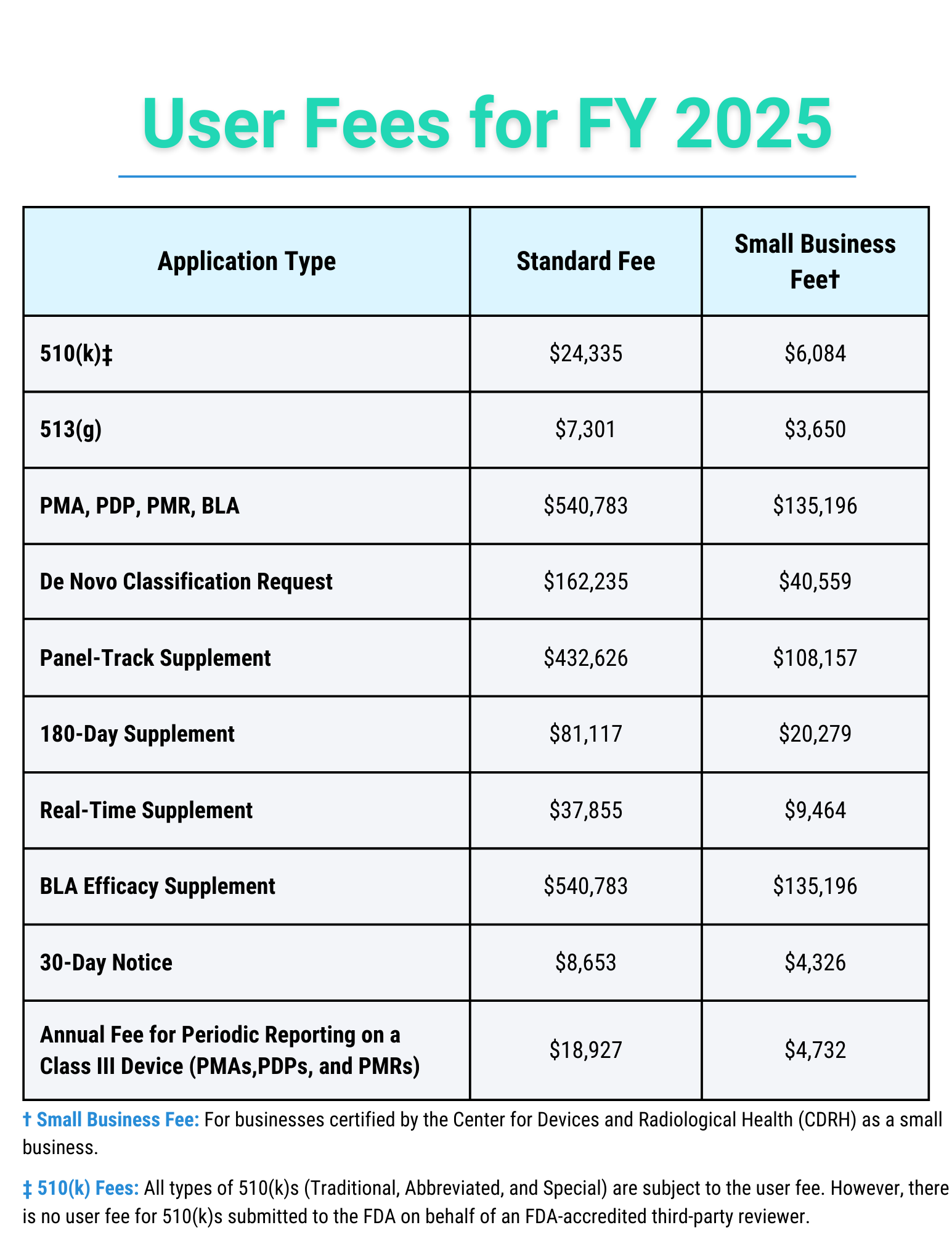
For more information on the user fees for FY 2025, click here.
Annual Establishment Fee
Under the Medical Device User Fee Amendment (MDUFA), all medical device manufacturers must pay the establishment registration fee, with no waivers or discounts given to small organizations. For FY 2025 (October 1, 2024 – September 30, 2025) the Annual Establishment Registration Fee is $9,280. The annual establishment fee is typically paid once the device has received clearance (30 days after the device is on the market).
What To Know Before You Start
Electronic Submissions To The FDA
The FDA guidance document, “Providing Regulatory Submissions for Medical Devices in Electronic Format – Submissions Under 745A(b) of the Federal Food, Drug, and Cosmetic Act” provides insight into the FDA’s process of developing templates to enable the preparation, submission, and review of electronic medical device regulatory submissions. The framework stated in the above guidance led to the development of the eSTAR templates. Since the development of the eSTAR templates, the FDA has published final guidelines for electronic submissions in their guidance, “Electronic Submission Template for Medical Device 510(k) Submissions.” This guidance provides information on the process of electronically submitting 510(k) submissions, information to include in your submission, and criteria for waivers and exemptions. The FDA published this guidance with the intention of improving submission consistency and escalating the review process.
Information Required
The FDA describes their expectations on what to include in your electronic submission in the guidance titled, “Electronic Submission Template for Medical Device 510(k) Submissions.”
The information below presents a summary of the current electronic submission template structure for 510(k)s, including an outline of the expected submission content that the submitter is required to provide in each section.
Submission Type
- Identification of key information that may be useful to FDA in the initial processing and review of the 510(k) submission, including content from current Form FDA 3514, Section A.
Cover Letter / Letters of Reference
- Attach a cover letter and any documents that refer to other submissions.
Applicant Information
- Information on the applicant and correspondent, if applicable, consistent with content from current Form FDA 3514, Sections B and C.
Pre-Submission Correspondence and Previous Regulatory Interaction
- Information on prior submissions for the same device included in the current submission, such as submission numbers for a prior not substantially equivalent (NSE) determination, prior deleted or withdrawn 510(k), Q-Submission, Investigational Device Exemption (IDE) application, premarket approval (PMA) application, humanitarian device exemption (HDE) application, or De Novo classification request.
Consensus Standards
- Identification of voluntary consensus standard(s) used, if applicable. This includes both FDA-recognized and non recognized consensus standards.
Device Description
- Identification of listing number if listed with FDA.
- Descriptive information for the device, including a description of the technological characteristics of the device including materials, design, energy source, and other device features, as defined in section 513(i)(1)(B) of the FD&C Act and 21 CFR 807.100(b)(2)(ii)(A).
- Descriptive information also includes a description of the principle of operation for achieving the intended effect and the proposed conditions of use, such as surgical technique for implants; anatomical location of use; user interface; how the device interacts with other devices; and/or how the device interacts with the patient.
- AI-Specific Considerations: If the device includes AI-enabled software, describe:
- The role of AI in the device’s functionality, including the specific intended use with an explanation of how AI is used to achieve the device’s intended use, as well as a description of intended users and use environment.
- Pre-determined Change Control Plan (PCCP) – Clearly define the specific, pre-specified changes anticipated post-market, outline the methodology for implementing modifications, and outline how the modifications align with the original indications for use.
- Indicate whether the device has a locked or adaptive AI model and summarize the types of modifications covered under the PCCP.
- Mechanisms in place to ensure reliability, repeatability, and mitigation of potential biases.
- For more detailed information, review the FDA draft guidance on marketing submission recommendations for AI-enabled device software functions.
- Information on whether the device is intended to be marketed with accessories.
- Identification of any applicable device-specific guidance document(s) or special controls for the device type as provided in a special controls document (or alternative measures identified that provide at least an equivalent assurance of safety and effectiveness) or in a device-specific classification regulation, and/or performance standards.
Proposed Indications for Uses (Form FDA 3881)
- Identification of the proposed indications for use of the device.
- The term indications for use, as defined in 21 CFR 814.20(b)(3)(i), describes the disease or condition the device will diagnose, treat, prevent,cure, or mitigate, including a description of the patient population for which the device is intended.
Classification
- Identification of the classification regulation number that seems most appropriate for the subject device, as applicable.
Predicates and Substantial Equivalence
- Identification of a predicate device (e.g., 510(k) number, De Novo number, reclassified PMA number, classification regulation reference, if exempt and limitations to exemption are exceeded, or statement that the predicate is a pre amendments device).
- The submission should include a comparison of the predicate and subject device and a discussion why any differences between the subject and predicate do not impact safety and effectiveness.
- For more information on the FDA’s decision-making process in determining substantial equivalence review, “The 510(k) Program: Evaluating Substantial Equivalence in Premarket Notifications.”
- A reference device should also be included in the discussion, if applicable.
Design /Special Controls, Risk to Health, and Mitigation Measures
- Applicable to Special 510(k) submissions only.
- Identification of the device changes and the risk analysis method(s) used to assess the impact of the change(s) on the device and the results of the analysis.
- Risk control measures to mitigate identified risks (e.g., labeling, verification).
- To learn more, review the Special 510(k) Program guidance document.
Labeling
- Submission of proposed labeling in sufficient detail to satisfy the requirements of 21 CFR 807.87(e).
- Generally, if the device is an in vitro diagnostic device, the labeling must also satisfy the requirements of 21 CFR 809.10.
- The term “labeling” generally includes the device label, instructions for use, and any patient labeling.
- AI-Specific Considerations: If the device includes AI-enabled software, describe:
- State that AI is used and explain its role in the device’s intended use.
- Summarize model inputs along with define model outputs and its intended use.
- Describe automation level with a high-level overview of the model architecture, data sources, demographics, and validation standards.
- Highlight performance metrics, offer installation instructions and system integration guidance.
- Provide patient-friendly labeling with instructions, risks, and limitations.
Reprocessing
- Information for assessing the reprocessing validation and labeling, if applicable.
- See the FDA guidance document, “Reprocessing Medical Devices in Health Care Settings Validation Methods and Labeling,” for more information on reprocessing validation and labeling requirements.
Sterility
- Information on sterility and validation methods, if applicable.
- For more information, review the FDA guidance, “Submission and Review of Sterility Information in Premarket Notification (510(k)) Submissions for Devices Labeled as Sterile.”
Shelf Life
- Summary of methods used to establish that device performance is maintained for the entirety of the proposed shelf-life (e.g., mechanical properties, coating integrity, pH, osmolality), if applicable.
- For details on the FDA’s regulations and policies relating to shelf life, see the guidance document, “Shelf Life of Medical Devices.”
Biocompatibility
- Information on the biocompatibility assessment of patient contacting materials, if applicable.
- Find further details on the FDA’s expectations for the biological evaluation of medical devices, here.
Software / Firmware
- Submission of applicable software documentation, if applicable.
- AI-Specific Considerations:
- Describe how the model was trained, if tuning evaluation was conducted, explanation of any pre-trained models that were used, quality control criteria or algorithms, and methods applied to the input and/or output data.
- The device’s user interface with an overview of the operational sequence of the device and the user’s expected interactions with the user interface.
- The AI model type, training methodology, and dataset characteristics (e.g., sources, diversity, representativeness).
- Adaptability features, including whether the model can change over time and how updates are managed by users in order for the device to maintain performance.
- Provide a description of workflows and user interactions with examples of outputs and recorded demonstration of the device.
- Include a Risk Management File with a detailed plan and risk assessment covering the entire device lifecycle, addressing risks related to user understanding and providing explanations of risk controls.
Cybersecurity / Interoperability
- Submission of applicable information regarding the assessment of cybersecurity, if applicable.
- For details on cybersecurity device design, labeling, and documentation requirements, see the FDA guidance, “Cybersecurity in Medical Devices: Quality System Considerations and Content of Premarket Submissions.”
- For more information on design considerations for interoperable medical devices, review the FDA guidance, “Design Considerations and Pre-market Submission Recommendations for Interoperable Medical Devices.”
- AI-Specific Considerations:
- Include unique considerations related to AI cybersecurity, providing evidence of appropriate cybersecurity testing for AI-specific risks and describe measures to address data vulnerabilities along with methods to address AI-specific cyber threats.
Electromagnetic Compatibility (EMC), Electrical, Mechanical, Wireless and Thermal Safety
- Submission of the EMC, Electrical, Mechanical, Wireless and Thermal Safety testing for your device or summarize why testing is not needed.
- Specific details on testing recommendations for EMC of medical devices can be found in the guidance document, “Electromagnetic Compatibility (EMC) of Medical Devices.”
- For information on specific considerations for radio frequency (RF) wireless technology in medical devices, see the FDA RF wireless guidance.
Performance Testing
- For non-in vitro diagnostic devices: Provide information on the non-clinical and clinical test reports submitted, referenced, or relied on in the 510(k) for a determination of substantial equivalence.
- Further information on testing report requirements can be found in the FDA guidance, “Recommended Content and Format of Non-Clinical Bench Performance Testing Information in Premarket Submissions.”
- For in vitro diagnostic devices: Provide analytical performance, comparison studies, reference range/expected values, and clinical study information.
- AI-Specific Considerations:
- Provide information on data collection, development, and test data independence, reference standards, and representativeness.
- Include information on performance validation testing used to assess model performance using independent data sets and include comprehensive study protocols detailing study design and study results.
References
- Inclusion of any literature references, if applicable.
Administrative Documentation
- Inclusion of additional administrative forms applicable to the submission, including but not limited to a general summary of submission/executive summary (recommended), a Truthful and Accuracy Statement, and a 510(k) Summary or statement.
- AI-Specific Considerations: AI-enabled software function devices should include the following in their “Public Submission Summary.”
- Include a clear declaration that AI is used in the device and explain its role as part of the device’s intended use.
- Describe the class of AI model and its limitations within the device.
- Provide statistical confidence levels for predictions and outline plans for updating and maintaining the AI model.
Amendment / Additional Information (AI) Response
- Inclusion of responses to Additional Information Requests.
The information above outlines the required content for your electronic 510(k) submission. All of these requirements are integrated directly into the eSTAR template.
As a result, you do not need to separately provide the following forms as part of your submission:
- Indications for Use Page (Form FDA 3881)
- Premarket Review Submission Cover Sheet (Form FDA 3514)
- Declaration of Conformity, If Applicable
For information on the FDA’s recommended documentation for AI-enabled devices, refer to the agency’s draft guidance titled, “Artificial Intelligence-Enabled Device Software Functions: Lifecycle Management and Marketing Submission Recommendations.”
eSTAR Versions
Please refer to the table below to identify the appropriate eSTAR PDF template for your specific submission type.
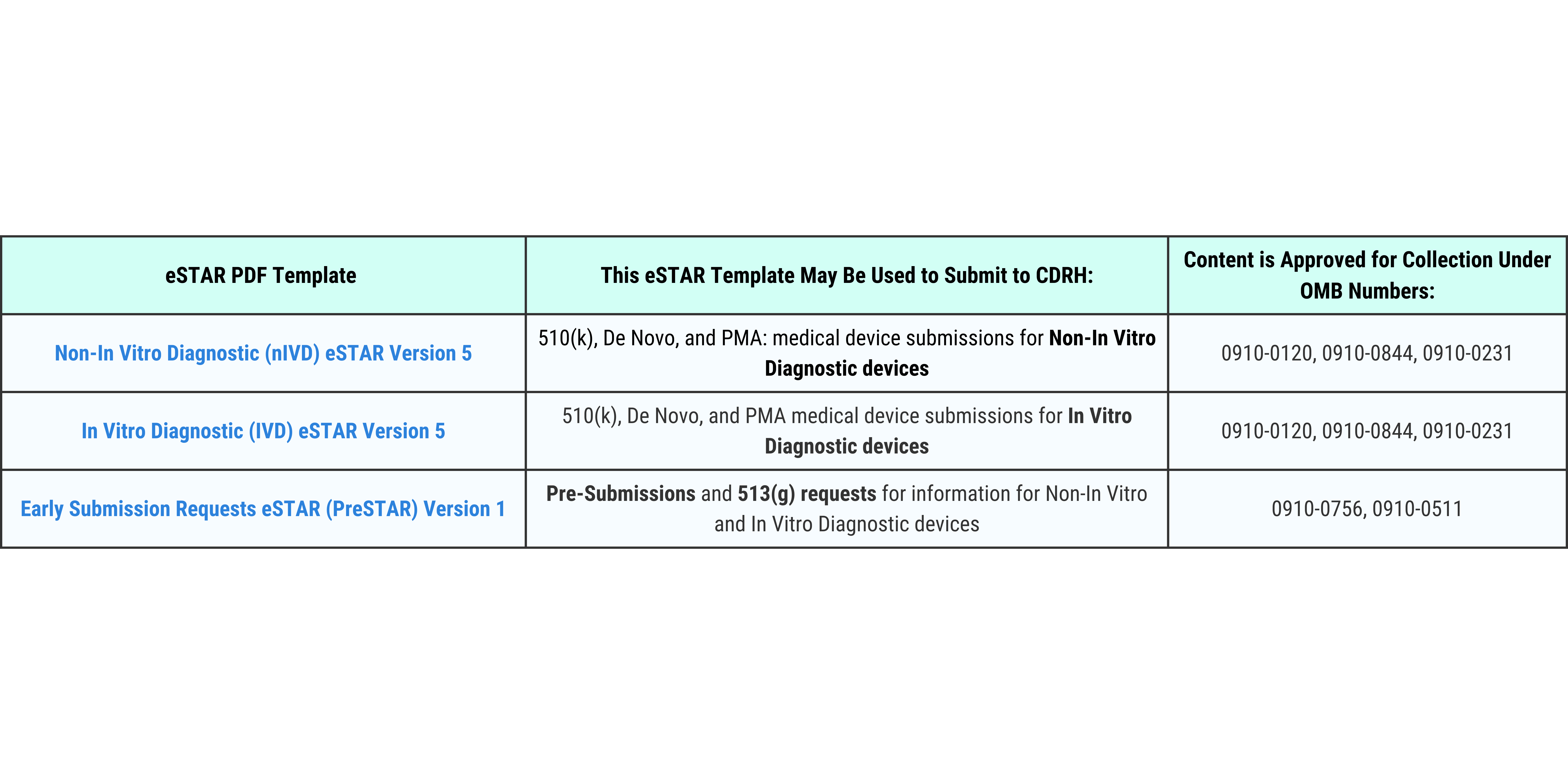 Access the template for your submission type by clicking the corresponding template name below:
Access the template for your submission type by clicking the corresponding template name below:
Non-In Vitro Diagnostic (nIVD) eSTAR Version 5
In Vitro Diagnostic (IVD) eSTAR Version 5
Early Submission Requests eSTAR (PreSTAR) Version 1
See download instructions in the next section (Preparation Steps).
For more details on what happens if a file version changes, using an agency for eSTAR submissions, or voluntary PreSTAR or 513(g) submissions, visit the FDA’s website.
Preparation Steps

The eSTAR template is an interactive PDF that is offered in three template versions: Non-In Vitro Diagnostic eSTAR Version 5, In Vitro Diagnostic (IVD) eSTAR Version 5, and Early Submission Requests eSTAR (PreSTAR) Version 1.
Select the template that aligns with your device type (for example, if your device is a COVID-19 detection device you would select the “In Vitro Diagnostic (IVD)” eSTAR template).Once you have selected your template, right click to save the PDF file before opening in Adobe Acrobat Pro or Adobe Acrobat Reader. The eSTAR PDF template cannot be opened in a web browser and must be downloaded prior to filling out.

Thoroughly read the instructions listed below or on the FDA’s website to ensure you have a good understanding of the eSTAR submission requirements and process.

Once you have downloaded the correct eSTAR PDF template and thoroughly reviewed the submission directions, begin filling out the template accordingly. The interactive PDF is designed to guide you through the necessary steps of the submission process, ensuring you provide comprehensive data for your premarket review.
File Size
It is important to note that the CDRH Portal cannot accept eSTAR submissions that are larger than 4 GB total or that have attachments greater than 1 GB. To submit an eSTAR that exceeds the requirements of the CDRH Portal, you will need to mail the electronic version of your submission to the CDRH Document Control Center (DCC). Mail your document to the below address:
U.S. Food and Drug Administration
Center for Devices and Radiological Health
Document Control Center (DCC) – WO66-G609
10903 New Hampshire Avenue
Silver Spring, MD 20993-0002
For additional information on mailing your eSTAR submission due to exceeding the file size limit, review the FDA’s eCopy Medical Device Submissions.
File Attachments
A variety of attachment types are accepted by eSTAR. Attachment types that are not accepted will be automatically prevented with a pop up explanation message.
You can sort your attachments in the attachment pane.
When possible, the FDA recommends combining attachments of related content (for example, Software Requirements Specifications). Combining attachments of similar content, will allow for less attachments to be needed in your eSTAR. To combine your attachments in Adobe Acrobat Pro, you can select “Tools” then “Combine Files” or combine them in a PDF Portfolio.
Completing Your eSTAR
Using The eSTAR Template
The eSTAR template is an interactive PDF that guides users through the submission process and is designed to enhance the quality of regulatory submissions to the FDA. The structured format of eSTAR promotes consistency across sections and the built-in validation tools automatically identify missing or incorrect information, helping provide comprehensive data that meets the FDA’s submission requirements. The eSTAR template also provides helpful instructions and interactive prompts to assist submitters in providing the required information.
Now that we’ve discussed some key aspects of the eSTAR templates, let’s discuss a few areas on the template that you should know about before filling out your eSTAR template.
Key Aspects of eSTAR Template
Significance of Color Coding
The eSTAR template uses the below color coding to let the submitter know the status of a question and what information is still needed for their submission. The color coding is shown throughout the eSTAR template on the left hand side of the document.

The below image shows an example of a section (Application Purpose) that has been successfully completed and a section (Application Sub-Type) that is missing required information.

Interactive Prompts and Instructions
A unique feature to the eSTAR template is the ability to access detailed information for each section in the application. Throughout the template there are interactive blue banners and question marks that provide the user with additional information once clicked on.
When clicking on the blue banners, you will be provided regulatory information pertaining to that question or section. In the below example, you can see the blue banner on the template says, “Show Application Introduction.” Once the banner has been clicked on, you can see there is detailed information provided on the submission types. Once you are done reading the displayed information, you can click “Hide Application Type” to no longer see the information in the drop down.
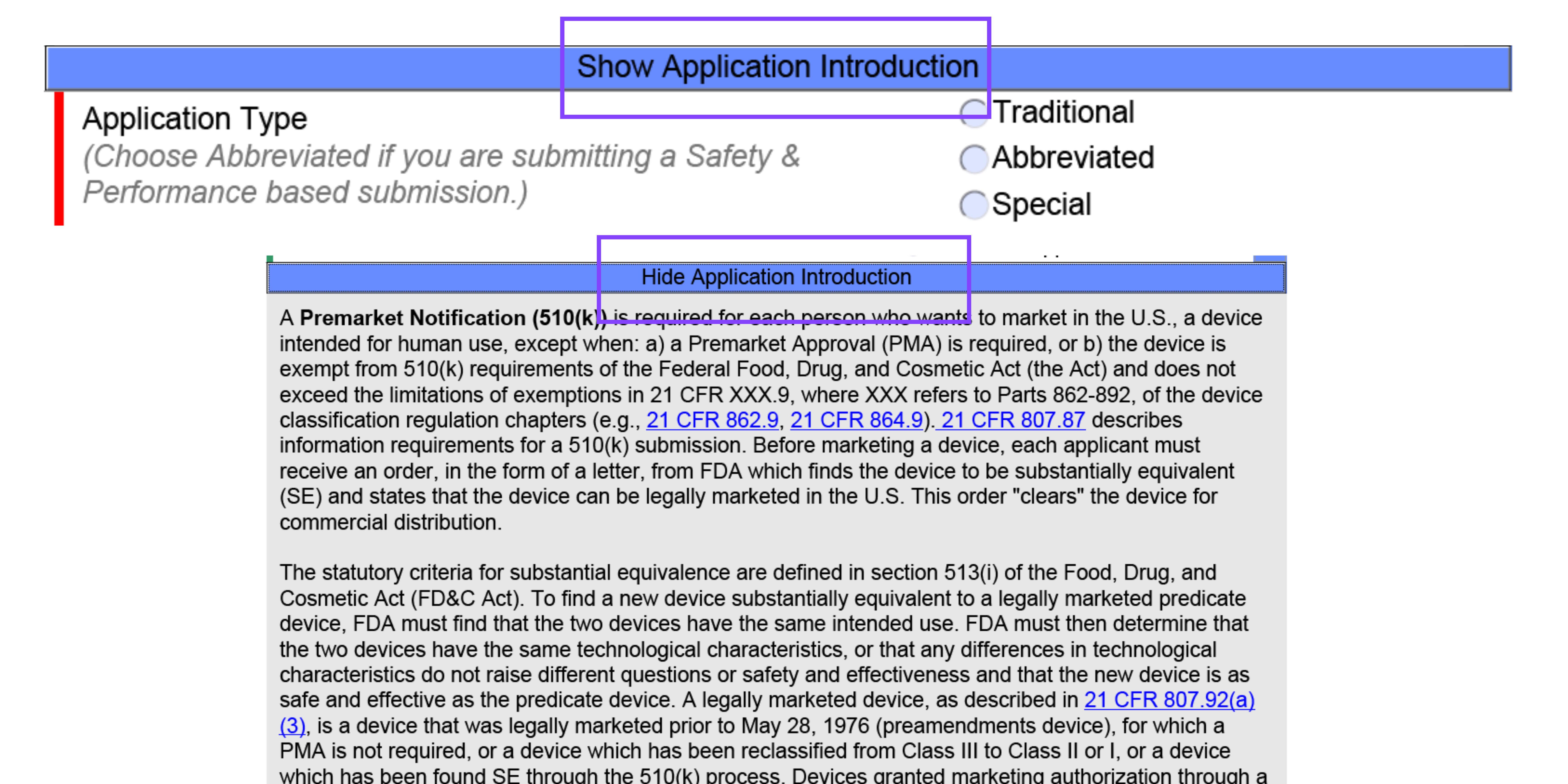
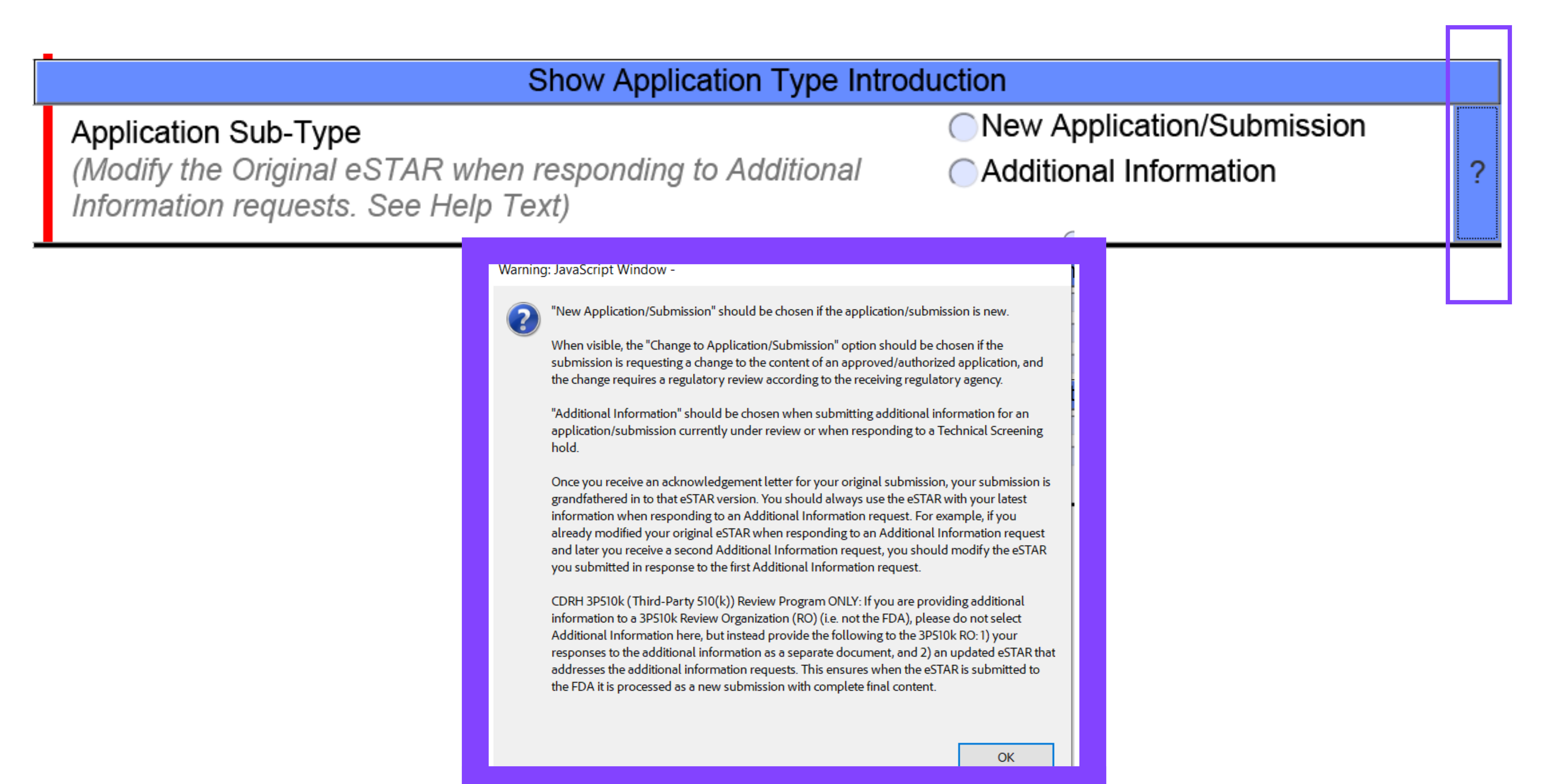
The blue question marks provide the user with context on the FDA’s expectations for that section, details behind the selection options, and further insight on what not to select based on various criteria stated. This feature of eSTAR gives users the ability to make educated decisions while gaining insight to the FDA’s thoughts pertaining to the correlated section.
In the below example you can see that the blue question mark is located on the right hand side of the template. Once clicked on, information on the answer choices for that section is displayed. Once you are finished reading the provided content, you can select “OK” to exit out of the pop up.
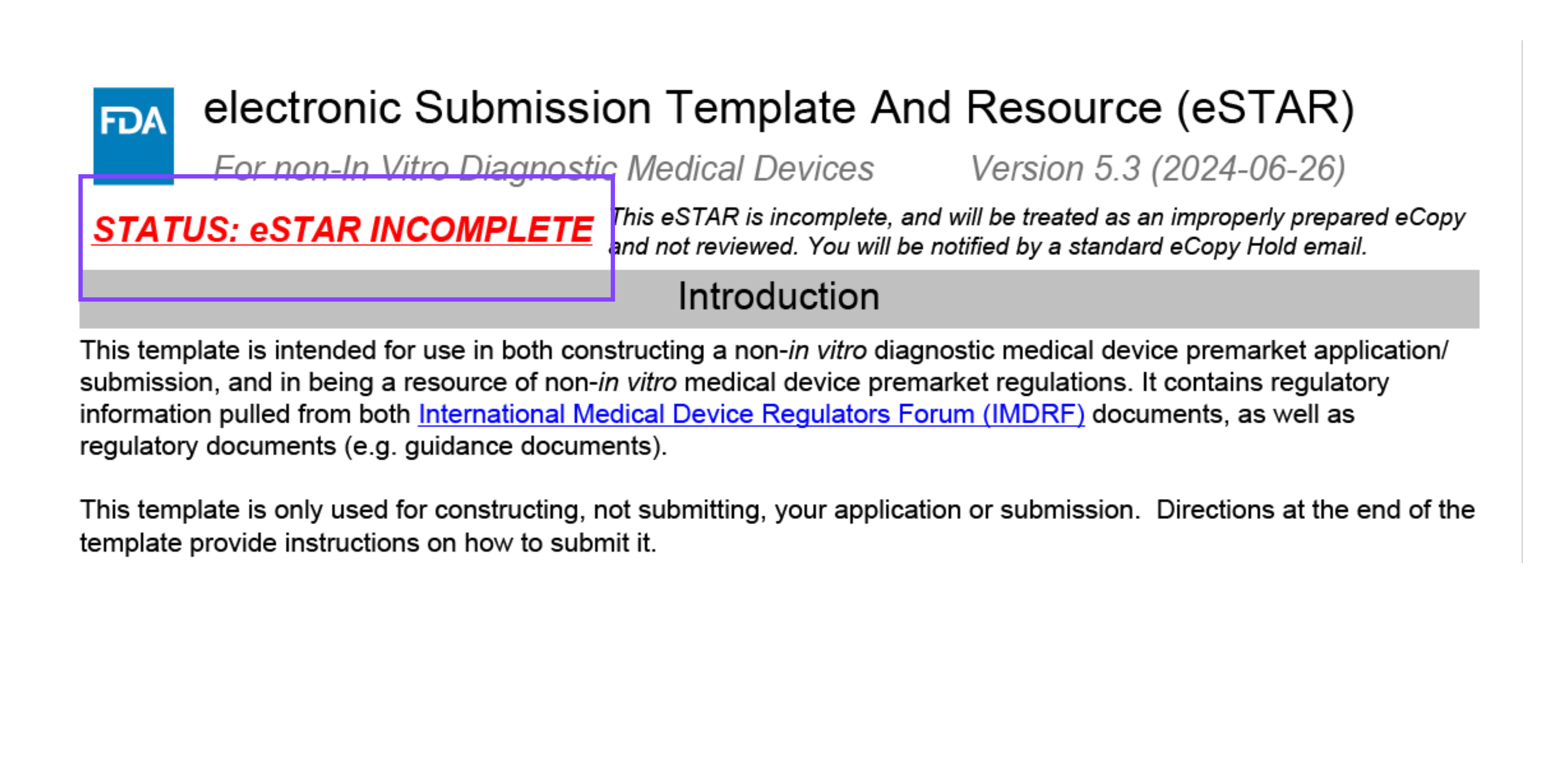
eSTAR Status Message
At the top of the first page on the eSTAR template, a status message will appear saying “STATUS: eSTAR COMPLETE” or “STATUS: eSTAR INCOMPLETE.” Due to the interactive nature of the eSTAR PDF template, the status message will reflect the progress made in your application. If all requirements in the submission application have been fulfilled then the status message will reflect this and will show “eSTAR COMPLETE.” If all requirements of your eSTAR have not been fulfilled, the status message will say, “eSTAR INCOMPLETE.” This is a great feature of eSTAR that allows users to track their progress when filling out their application.
Submitting an eSTAR
The FDA requires all 510(k) submissions, unless exempted, to submit their electronic submission using eSTAR through the CDRH Portal or through the Electronic Submission Gateway (ESG) for CBER.
Submitting Through The CDRH Portal
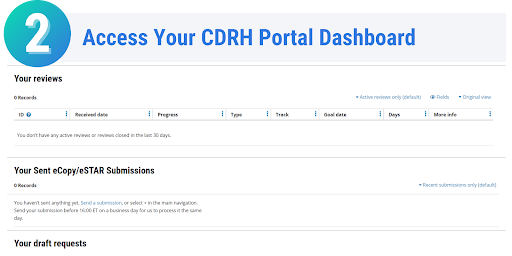
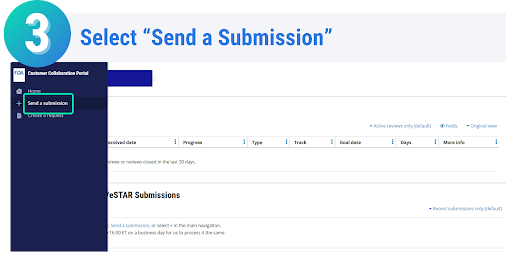

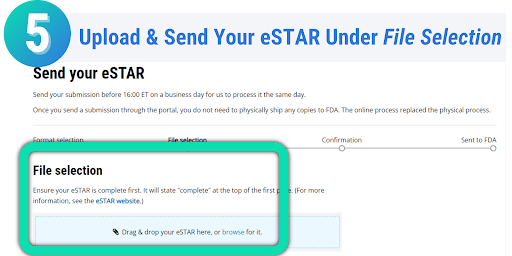
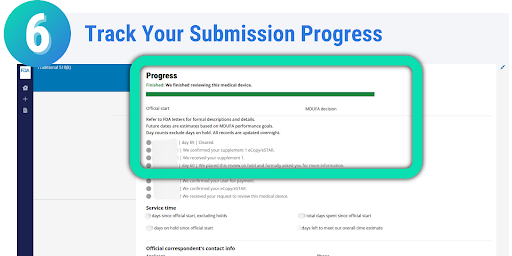
Once your eSTAR has been submitted, you can track the progress of your submission through the review process. On the Progress page in the CDRH Portal, messages will be displayed on when your eSTAR has been received, when it is being reviewed, if additional information is needed, and the overall status of your submission. Each message displayed on the progress page is stamped with the date, allowing you to easily monitor the progress and status of your submission
Submitting Voluntary eSTARs
For details on where to submit your voluntary eSTAR submission per your submission type, see below:
De Novo: CDRH Portal or Mail to FDA
Pre-Sub Submissions: CDRH Portal or Mail to FDA
PMA: CDRH Portal or Mail to FDA
513(g) Requests: CDRH Portal, CBER’s Electronic Submission Gateway(ESG), or Mail to FDA
FDA Mailing Addresses:
De Novo, Pre-Subs, and PMA eSTARs submitted by mail should be sent to the CDRH’s Document Control Center (DCC):
U.S. Food and Drug Administration
Center for Devices and Radiological Health
Document Control Center (DCC) – WO66-G609
10903 New Hampshire Avenue
Silver Spring, MD 20993-0002
513(g) eSTARs submitted by mail should be sent to CBER’s Document Control Center (DCC):
U.S. Food and Drug Administration
Center for Biologics Evaluation and Research
Document Control Center
10903 New Hampshire Avenue – WO71, G112
Silver Spring, MD 20993-0002
Review Process
Review Process and Timeline
The FDA’s review timeline for an eSTAR submission follows the same general framework as traditional 510(k) submissions but benefits from increased efficiency due to the standardized electronic format. The specific timeline for review varies depending on the device type and the complexity of the submission.
The below diagram represents the typical review process.
Once the FDA receives an eSTAR submission, it is expected that, since the electronic submission is properly prepared using the electronic submission template, it should be considered complete. Therefore, eSTAR submissions are not expected to go through the refuse to accept (RTA) process. However, the FDA will still conduct virus scanning and a technical screening of the eSTAR submission.
Technical Screening
- Following the initial submission, the FDA typically requires 1-2 weeks to perform a technical screening. If the device does not pass this screening, the FDA will inform the submitter via email about the missing or incomplete information. The 510(k) process will be paused until a revised submission is provided. If the updated submission is not received within 180 days of the notification, the FDA will consider the 510(k) application withdrawn.
Review Phase
- In the review phase, the FDA thoroughly evaluates the submission, examining clinical data, product quality, and manufacturing processes to ensure the device meets safety and efficacy standards. Depending on the complexity of the submission, this process may take 6 to 12 months. During this time, the FDA may request additional information or clarifications from the submitter. It is essential for stakeholders to respond promptly to these requests to prevent any delays and ensure the review process stays on track. Delays in responses can extend the overall timeline and impact the final decision.
Decision Phase
- The final decision on an eSTAR submission may take anywhere from 6 to 18 months following the initial submission, depending on the complexity of the device and the completeness of the application. The FDA’s decision could result in full approval, conditional approval (which may require the submission of additional data or clarifications), or outright rejection. Various factors, such as the need for further review or additional testing, could impact the timeline. It is also important for submitters to respond promptly to any requests for Additional Information (AI) from the FDA to avoid delays in the decision-making process.
The rest of the 510(k) review will proceed in accordance with the FDA’s guidance “The 510(k) Program: Evaluating Substantial Equivalence in Premarket Notifications [510(k)],” and will follow the protocols outlined in 21 CFR 807 subpart E.
Reviewing Mailed Submissions
If a mailed eSTAR submission is incomplete, the FDA will notify the submitter via email, detailing the missing information. The submission will then be put on hold for up to 180 days or until a fully revised eSTAR is resubmitted to the FDA.
De Novo Requests
A De Novo classification request will be reviewed in accordance with 21 CFR part 860, subpart D, as outlined in the FDA guidance document, “De Novo Classification Process (Evaluation of Automatic Class III Designation).”
PMA Submissions
A PMA submission will be evaluated based on the regulations specified in 21 CFR part 814.
Benefits and Challenges of eSTAR
Benefits of Using eSTAR
eSTAR provides applicants with an interactive PDF template designed by the FDA, guiding applicants through the medical device submission process ensuring they provide all necessary information. The FDA created the eSTAR program with the intention of enhancing the quality of submissions, while streamlining the regulatory submission process. eSTAR offers users the following benefits:
Guided Submission Process
- eSTAR helps submitters ensure they include all required details for a complete submission, reducing errors and omissions.
Alignment with FDA Review Tools
- It complements the reviewers’ internal Submission Memo And Review Template (SMART), ensuring that the information provided matches what the reviewer expects.
Standardized Format
- The standardized format makes information easily accessible for both the reviewer and submitter, streamlining communication and clarity.
Automation and Efficiency
- By automating many aspects of the submission process, eSTAR ensures that all necessary content is present, eliminating the need for a Refuse to Accept (RTA) review and avoiding RTA holds.
Auto-fill Functionality
- It automatically fills entered information to prevent redundant data entry, saving time for submitters.
Built-in Databases
- eSTAR integrates built-in databases to accurately auto-fill information related to device-specific guidances, classification identification, and standards.
Pre-loaded Forms
- It includes essential forms, such as the Truth & Accuracy statement, Form 3514, 510(k) Summary, Declaration of Conformity, and Indications for Use Form 3881, making it easier to complete the submission.
Structured Data Collection
- eSTAR collects submission data in a structured format, helping automate various aspects of FDA processing, enhancing efficiency.
Comprehensive Resource
- It consolidates all necessary information and links into one platform, serving as a valuable resource for submission preparation.
Potential Challenges of eSTAR
While eSTAR offers several advantages in streamlining the FDA submission process, it also presents some challenges for submitters, especially those new to the system. These challenges can affect the ease of use, preparation, and submission process. Below are a few common challenges of using eSTAR:
Learning Curve for New Users
- For organizations not familiar with the eSTAR system, there can be a significant learning curve in understanding and navigating the new submission format.
Technical Limitations
- Filling out the eSTAR template requires the use of Adobe Acrobat which limits document editing to one user at a time, which can result in the increased risk of errors especially with multiple teams contributing to the submission.
Limited Flexibility
- The structured nature of the eSTAR format may limit the ability to customize submissions for unique devices or circumstances, which can pose difficulties for complex submissions.
Dependence on Built-in Databases
- While eSTAR’s built-in databases help auto-fill information, outdated or incomplete data in these databases could lead to incorrect or incomplete submissions, causing delays.
System Updates and Changes
- Regular updates to the eSTAR tool may require users to stay up-to-date with new features or changes in functionality, which can add to the complexity of managing submissions over time.
Collectively, these challenges of eSTAR can result in an increased risk of submission rejection, creating a delay to market entry, if not handled correctly.
Overcoming These Challenges
To effectively address the challenges associated with using eSTAR, users can adopt several strategies to streamline their submission process and enhance their overall experience. Here are some recommendations:
Training and Resources
- Users should take advantage of training sessions, webinars, and instructional materials provided by the FDA or industry experts, such as Rook Quality Systems. Familiarizing themselves with the eSTAR system will help reduce the learning curve and improve confidence in navigating the platform.
Technical Support
- Establish a relationship with technical support teams who can assist with any compatibility issues or technical limitations encountered during the submission process. This proactive approach can help resolve problems quickly and efficiently.
Thorough Preparation
- Before starting the eSTAR submission, users should gather all necessary documentation and information in advance. Creating a checklist of required data and forms can help ensure completeness and accuracy, minimizing the risk of errors.
- Using a robust eQMS like QuickVault simplifies the submission process by keeping your documents organized and ensuring seamless version control. With QuickVault, you can easily track the latest document versions, improving efficiency and streamlining your submission workflow.
Utilize Templates and Examples
- Referencing completed eSTAR submissions or templates can provide valuable insights into formatting and required content. This can help users understand how to structure their submissions effectively.
Regular Database Reviews
- To mitigate the risk of using outdated information from built-in databases, users should regularly verify and update their data sources. This practice can ensure the accuracy of the information being submitted.
Seek Feedback and Collaboration
- Partner with a consultancy like Rook Quality Systems to help put together your eSTAR or review the submission before submitting to the FDA. Having regulatory experts who are proficient in eSTARs and the FDA’s expectations can help you identify arrears of improvement and enhance the overall quality of your submission.
Stay Informed on Updates
- Users should subscribe to FDA newsletters, alerts, or announcements regarding eSTAR updates. Being aware of any changes or new features can help users adapt quickly and leverage the latest tools available.
Patience and Flexibility
- Recognizing that challenges may arise is crucial. Users should remain patient and flexible in their approach, being willing to adapt their strategies as needed to overcome any obstacles encountered during the submission process.
By implementing these strategies, users can effectively navigate the potential challenges of eSTAR and streamline their submissions to the FDA.
Achieving Submission Success
Tips for Submission Success
We’ve discussed the benefits of eSTAR, the challenges that can arise from using eSTAR, and how to overcome these challenges but now let’s explore the best practices for submission success.
At Rook Quality Systems, we’ve gained extensive experience through the development, review, and submission of numerous eSTARs. This hands-on expertise has given us a deep understanding of both the advantages and potential hurdles that come with using the eSTAR system. Drawing from our experience, we’ve compiled some valuable tips to help ensure your eSTAR submission is both efficient and successful.
- Utilize Templates – Begin by creating copies of the eSTAR templates for your internal use. This practice allows for more efficient collaboration among team members, making it easier to gather and input the required information collectively.
- Centralized Document Control – QuickVault allows you to manage all your submission-related documents in one secure, cloud-based platform. This centralization ensures that your team always has access to the latest versions, minimizing errors and improving collaboration.
- Implement Secure File Storage – Utilize secure, cloud-based storage solutions to manage eSTAR submissions. Given the variety, size, and potential sensitivity of the files involved, it is crucial to maintain a centralized location with controlled access to protect sensitive information and facilitate easy retrieval.
- Automated Version Tracking – With QuickVault, you can easily track document changes and maintain full version control, ensuring that the most up-to-date documents are used in your submission, reducing the risk of compliance issues.
- Effective Planning – Develop clear checklists and timelines to assign specific tasks within the eSTAR process to the appropriate departments. This approach helps ensure accountability, keeps track of deadlines, and ensures that the necessary work is completed. Since the eSTAR process can take several days for completion and approval, it’s important to allocate extra time to accommodate any unexpected delays.
- Invest in Training and Standard Operating Procedures – Allocate at least two weeks for training each time there is a regulatory change within the eSTAR framework. Since these changes happen frequently, it is advisable to consider engaging a consultancy like Rook Quality Systems to manage this training workload effectively. This approach can provide access to expertise and resources that may not be readily available internally. Additionally, collaborating with such a consultancy can help develop and update Standard Operating Procedures (SOPs) to ensure they align with the latest requirements and best practices.
- Foster Collaboration and Communication – Encourage open communication among all stakeholders involved in the submission process. Regular meetings or updates can help address any issues early and ensure everyone is aligned with their responsibilities.
- Conduct Mock Submissions – Performing practice submissions using the eSTAR templates can help identify potential challenges before the actual submission. This proactive approach allows teams to fine-tune their processes and become more familiar with the system.
- Monitor and Review – After the submission, it’s essential to review the entire process and gather feedback from all participants. Identifying what worked well and what could be improved will help refine future submissions and enhance efficiency.
By following these tips, you can enhance your chances of a successful eSTAR submission, ensuring compliance and streamlining the process for future applications.
Leveraging Industry Expert Support
Rook Quality Systems
Rook Quality Systems is dedicated to supporting startups to Fortune 500 medical device companies in the development and upkeep of robust quality systems, providing key resources and expert insight every step of the way. We offer comprehensive regulatory services, assisting companies in navigating FDA compliance, ISO certifications, and global market entry, ensuring your medical devices meet all necessary regulatory requirements.
Our mission is to enable our clients to implement compliant Quality Management Systems (QMS) to ensure that they can efficiently produce effective and reliable medical devices. We partner with our clients to ensure they have a robust regulatory and quality plan to get to market quickly and maintain compliance. RookQS consultants provide dedicated support for all aspects of regulatory compliance and quality management to ensure all medical devices are designed and manufactured with the highest quality.
Your eSTAR Specialists for Simplified, Seamless Submissions
Rook Quality Systems is committed to supporting clients through the entire eSTAR submission process. With our deep expertise in regulatory compliance and experience in navigating FDA requirements, we help ensure that your eSTAR submission is accurate, complete, and aligned with all necessary guidelines. Our team will guide you through the structured eSTAR template, providing expert input on device description, labeling, risk analysis, and clinical data, while leveraging eSTAR’s automated checks to reduce the chance of errors or omissions that could delay approval.
By partnering with Rook, you gain a dedicated team that will streamline your submission process, minimize documentation issues, and enhance your chances for a quicker review and approval. Whether you’re submitting a 510(k) or De Novo request, Look To Rook for regulatory expertise and tailored support to ensure that your eSTAR submission is well-organized, efficient, and ready for FDA evaluation.
QuickVault By Veeva Systems
QuickVault is a purpose-built electronic Quality Management System (eQMS) designed specifically for MedTech companies. Developed by Veeva Systems, the world’s leading provider of cloud-based solutions for the Life Sciences industry, QuickVault addresses the unique needs of smaller MedTech firms, supporting both pre-commercial technology development and scalable growth during commercial distribution.
QuickVault enhances cross-functional efficiency across the entire organization, offering robust functionality that serves R&D, Quality, Regulatory, and Operations teams. This broad approach makes QuickVault more than an eQMS – it is seen by the industry as a collaboration platform for MedTech efficiency and commercial success.
Streamlining FDA eSTAR Submissions with QuickVault
QuickVault automates and streamlines MedTech product development under Design Controls. As a result, many of the documents required for eSTAR are readily available on QuickVault in a structured manner. In addition, QuickVault’s document management features provide users with full transparency and control over document statuses, ensuring the correct and latest documents are included in submissions.
QuickVault’s document management features provide users with full transparency and control over document statuses, such as draft, effective, or in revision, which are crucial for completing submissions. The platform’s tagging system further enhances efficiency by allowing users to apply a “submission” tag. With this tag, all relevant documents for a specific submission can be instantly identified, sorted, and filtered with a single click, streamlining the entire process.
In 2025, Veeva will enhance QuickVault by introducing regulatory functionality, making it even easier to populate and track submissions like 510(k) and CE marking.
